9班
1.プリオン病とは?
プリオン病は、プリオンと命名された蛋白
(prion protein)が中枢神経に蓄積し、神経系を高度に荒廃させ、死に至らせる感染性の疾患です。代表的なものが、ヒトのクロイツフェルト・ヤコプ病ですが、動物でも、下記の表のように種々のものが知られており、人畜共通感染症として感染予防に関心が持たれています。
プリオン
(Prion)は、proteinaceous infectious proteinの略で、プリオン病の感染物質としてPruisnerによって名付けられました。感染性がありますが細菌、ウイルスなどの病原体ではなく、患者の自己蛋白です。ただし、患者の脳に蓄積しているプリオン蛋白は、正常の構造とは立体構造が異なるものです。この異常構造のプリオン蛋白が体内に入ると、その機序は未だよく判りませんが患者の脳にある正常構造のプリオン蛋白の構造を変え、長い潜伏期を経て発病させるに至ります。
人畜共通感染症とは
プリオン病の分類
|
特発性クロイツフェルト・ヤコブ病 |
||
|
孤発性 |
医原性クロイツフェルト・ヤコブ病 |
|
|
プリオン病 |
異型クロイツフェルト・ヤコブ病 |
|
|
ヒト |
kuru |
|
|
遺伝性 |
家族性クロイツフェルト・ヤコブ病 |
|
|
プリオン病 |
ゲルストマン・ストロイスラー・シャインカー病 |
|
|
家族性致死性不眠症 |
||
|
ヒツジ |
スクレイピー |
|
|
ヤギ |
||
|
オオシカ |
慢性るいそう病 |
|
|
ミンク |
伝達性ミンク脳症 |
|
|
ウシ |
牛海綿状脳症(狂牛病) |
|
|
ネコ |
猫海綿状脳症 |
|
|
トラ |
||
|
ピュ-マー |
||
|
チーター |
||
Kuru
(クールー)についてニューギニアの高地に住むフォア族に小脳失調による歩行障害が多発しているのが1957年に報告(Gajdusek & Zigas)されたのですが、患者は成人女子と小児に多く、4か月から2年で死亡していました。
当初、遺伝性疾患と考えられ、遺伝形式についても色々検討されましたが、KuruとScrapieの臨床所見、病理所見の類似性が指摘(Hadlow,1959年)され、Gajdusekら(1966)がKuruの患者脳をチンパンジーの脳内に摂取することにより18~30か月後にチンパンジーに臨床症状を発症させることに成功しました。続いて、彼らは継代摂取にも成功し、結局のところ、フォア族における葬儀の際の人食いの習慣(cannibalism)が明らかとなりました。そして、食人の習慣がなくなることにより、患者数が激減し、今日では新たな患者の発生はほとんどなくなりました。
Gajdusek
とGibbsはこのKuruとクロイツフェルト・ヤコプ病の病因に関する一連の研究成果によってノーベル賞を得ています。
スクレイピー(
Scrapie)18世紀頃より英国、西ヨーロッパで知られていた羊の病気で、症状は羊がしきりに身体を柵などにこすりつけ(scrapie)、次いで刺激過敏、振戦、後ろ脚の脱力が生じます。数カ月から数年以内に死亡する病気として、牧畜業者に恐れられていました。
Cuille & Chelle(1936)
が病気の羊の脳を他のヒツジ、ヤギ、マウスの脳に接種し、発病させることに成功し、感染性の疾患であることを証明しました。牛海綿状脳症(狂牛病)
1986年頃より英国で発生した牛のプリオン病で、スクレイピ-に似た臨床症状を生じます。これは、離乳期の子牛の発育を促進させるために飼料の中に混ぜた羊の肉、骨の粉末の中にスクレピーに感染していたものが混じっていたためにヒツジのスクレピーが牛に感染したものと推定されておりますが、これまでに130万頭の牛が発病したといわれています。
異型クロイツフェルト・ヤコプ病
1996
年3月にイギリス政府は、それまでの古典的なクロイツフェルト・ヤコプ病とは少し異なる型のものがあり、牛海綿状脳症(狂牛病)と関係があるかもしれないと発表し、食品衛生、薬物行政の上で大問題になりました。現在は狂牛病の発生地域からの乳製品(薬物を含めて)の輸入が禁止されております。
また、厚生省はクロイツフェルト・ヤコプ病に関する緊急調査研究班を組織し緊急に全国調査をしましたが、今のところ日本国内での発症者の報告はありません
2、プリオン蛋白質の構造と機能
プリオン蛋白質(プリオン)
・海綿状脳症(スクレイピー)の病原因子とされる蛋白質性感染因子。
・これには正常なものと、病原性・感染性を持つ異常なものが存在する。正常プリオン蛋白と異常プリオン蛋白の違いは蛋白の高次構造の違いで、正常プリオン蛋白はαヘリックス、異常プリオン蛋白はβシート状構造をとる。また、正常プリオン蛋白は合成時間が短く、細胞膜の上で急速に破壊されていく。異常プリオン蛋白は合成時間がはるかに長く、細胞内に蓄積して分解されにくい。
・正常プリオン蛋白は健常な動物にも存在し、異常プリオン蛋白だけがプリオン病を引き起こす。
・プリオン蛋白遺伝子は、第20番染色体上に存在し、蛋白は253個のアミノ酸より構成されている。脳に一番多く、他の臓器でも発現している。
・正常細胞は正常なプリオン蛋白を生産し、スクレイピーへの感染により感染型(異常)プリオン蛋白に変化して、細胞内外に蓄積する。
・プリオン蛋白遺伝子を欠いたノックアウトマウスでは、スクレイピー病原因子を接種しても発病しない。つまり、異常なプリオンが増殖してプリオン病を発症するためには、正常なプリオン遺伝子が存在することが必要である。しかし、異常プリオンがプリオン遺伝子を異常化するのではない。(プリオン病症例の9割ではプリオン遺伝子の異常は見られない。)
3、どのような医療行為によってクロイツフェルト・ヤコプ病が感染する可能性があるか?
クロイツフェルト・ヤコプ病は、感染性の疾患なので、医療行為によって感染することがあります。今までに報告のあるものとしては、
1
脳手術、脊髄手術に伴うヒト硬膜移植
脳手術の際に、欠損した硬膜を補うためにヒトの死体から取られた硬膜を処理して用いていましたが、その硬膜移植者よりクロイツフェルト・ヤコプ病の発病者がでています。クロイツフェルト・ヤコプ病に関する緊急調査研究班の集計では、1987年頃までの移植者での発症が多く、1997年3月末までに43例が報告されています。1997年3月に、ヒト硬膜の移植を禁止されました。
今後は、これらの移植者で未発病者(潜伏期の長さからみて、これから発病する可能性は少ないと思われますが)の再手術の際の感染の拡大防止(使用後の器具の消毒)が重要な課題となっています。
2 下垂体由来の生長ホルモン製剤
かって、成長ホルモンの製剤が、死亡したヒトの下垂体から抽出して作られていた時代に、下垂体を取り出した患者の中にクロイツフェルト・ヤコプ病患者が紛れ込んでいたために、その製剤を注射した子供に多数のクロイツフェルト・ヤコプ病の発症をみました。日本での発症報告はありません。1985年以後の成長ホルモンは遺伝子組み替えによって作られているので安全です。
3 角膜移植
クロイツフェルト・ヤコプ病患者からの角膜移植(移植後にドナーの診断が明らかになりました)によって発病したという報告があります。
4 脳深部電極による脳波記録
てんかん手術の際にクロイツフェルト・ヤコプ病患者に使用された脳深部電極が使用され、
痴呆のある患者、診断の明らかでない患者はクロイツフェルト・ヤコプ病である可能性が常にあり、それらの脳脊髄の手術に際しては、使用後の器具の再使用による感染防止の観点から
、また、術者の安全のためにも厳重な注意が必要とされています。また、血液製剤を介しての感染を防ぐために下記の方からの供血は避ける必要があります。
・血縁者にクロイツフェルト・ヤコプ病と診断された人がいる。
・下垂体由来の成長ホルモンの注射を受けたことがある。
・角膜移植を受けたことがある。
・脳脊髄の手術で硬膜移植を受けたことがある。
4、クロイツフェルト・ヤコプ病の脳ではどのような病理学的な変化がみられるか?
発病してから
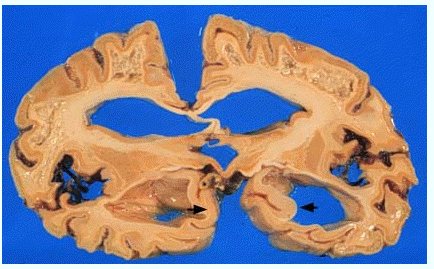
2年で亡くなられた患者さんの脳です。大脳の前額断面
(国立療養所犀潟病院神経病理 巻渕隆夫先生提供)
脳重
(750g)は正常(1200g)のほぼ半分に減少しています。大脳の皮質と白質は強く萎縮し、脳室は拡大しています。
しかし、大脳辺縁系特に海馬(矢印)は他の類似した疾患(無酸素脳症や低血糖脳症など)に比べて比較的軽度であるのが特徴です。
早期の例では萎縮が軽度の場合もあります。
小脳と延髄の水平横断面
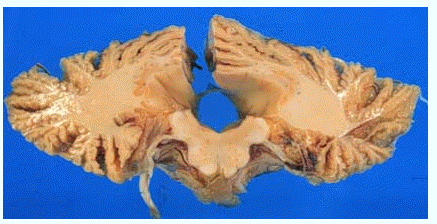
(国立療養所犀潟病院神経病理 巻渕隆夫先生提供)
小脳皮質は強く萎縮しています。
第4脳室も拡大しています。
大脳皮質の顕微鏡弱拡大像(標本は
HE染色)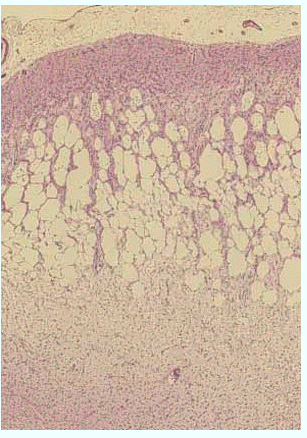
(国立療養所犀潟病院神経病理 巻渕隆夫先生提供)
萎縮した大脳皮質を弱拡大で見ると、大脳皮質特に中層から深層にかけ海綿状になっています。
そのためこの病気は海綿状脳症とも呼ばれます。
更に拡大を上げて見ると、神経細胞は変性消失し、強い星膠細胞の増殖(グリオーシス)が見られます。
5、クロイツフェルト・ヤコプ病の患者を介護する際の感染の危険性について
CJD
患者の感染性について
クロイツフェルトヤコプ病の疫学調査では、家族発生の例はほとんどなく、気道や消化管からの感染は考えにくいとされています。したがって、
通常の状態での接触感染の証拠はなく、不必要な恐怖を持つ必要はありません。しかし、角膜移植、硬膜移植やヒト脳下垂体から抽出した成長ホルモンの注射などで病気が伝播されたと言う報告があり、血液などのついた器具の取り扱いには充分な注意を要します。
感染防止ための注意点
1
通常のウイルスに対して行われている消毒法(紫外線、エタノール、ガス滅菌、オー トクレーブなど)は無効です。病理組織はホルマリン固定後にも感染力を失いません。
2
患者に接する医療従事者、家族は、手の汚染(傷口からの感染)、針刺し事故、感染物の眼への飛沫などに注意が必要です。(CJD患者の採血、髄液採取、口腔の清拭時の咬傷や爪による引っかき傷、汚染した手で目をこするなどに最大限の注意が必要です)
3
CJD患者に使用した手術器具は、出来るだけデイスポーザブル製品を使用し、汚染したものは焼却するようにします。
組織別の感染性の強さの分類
(WHOによる)
第一類
(強い感染性)
脳、脊髄
第二類
(中等度の感染性)
脾臓、扁桃腺、リンパ節、回腸、近位結腸
第三類
(弱い感染性)
坐骨神経、下垂体、副腎、遠位結腸、鼻粘膜
(非常に弱い感染性)
脳脊髄液、胸腺、骨髄、肝臓、肺、膵臓
第四類
(感染性が検出できない)
骨格筋、心臓、乳腺、乳、血餅、血清、便、腎臓、甲状腺、唾液腺、唾液、卵巣、子宮、睾丸、精嚢
以上のようなことから、
患者への対応としては
1
原則として、個室の必要はないが、吐血、下血、下痢、気道感染症などのあるときには個室が必要となる場合がある。
2
家族などの面会の制限の必要はない。
3
診察用具は専用にし、定期的に洗浄、消毒する。
4
衣類は汚染されていないものは通常の洗濯でよいが、血液、体液で汚染されたものは焼却、あるいはオートクレーブ滅菌後、洗濯する。
5
尿や吸引した喀痰は、容器の中に水酸化ナトリウム顆粒を1規定になるように投入し、1時間後に汚染槽に流す。使用済みの紙おむつは焼却する。
6
床が血液、体液などで汚染されたときには、水酸化ナトリウム溶液、または次亜塩素酸ソーダで洗浄する。
--------------------------------------------------------------------------------
6、クロイツフェルト・ヤコプ病ではどんな症状がみられるか?
抑うつ、不眠、記憶・判断力の低下、行動異常など漠然とした前駆症状の後、急速に痴呆が進行するとともに、四肢の筋力低下と硬直、振戦、あるいは小脳症状による歩行障害などが生じてきます。特徴的なのは、音や皮膚に触れるなどの刺激に対する感受性の亢進とミオクローヌス(ちょっとした音に対してもピクッと身体をふるわせるなど)です。そして、これらの症状は急速に進行し、精神荒廃、除脳硬直(手足を伸ばしたまま硬くなって、ちょっとした刺激によっても手足を強く伸展する動きを示す状態)、昏睡状態となり、死に至ります。全経過は数カ月、長くて
2年以内(平均17か月)のことが多いようです。
--------------------------------------------------------------------------------
7、クロイツフェルト・ヤコプ病の診断はどうするのか?
脳波所見
臨床的なミオクローヌスに一致して、脳波で周期性同期性放電(PSDという)がみられます。
周期性同期性放電は、0.5~2秒間隔でくり返す棘波あるいは徐波の周期的な波形をいいます。
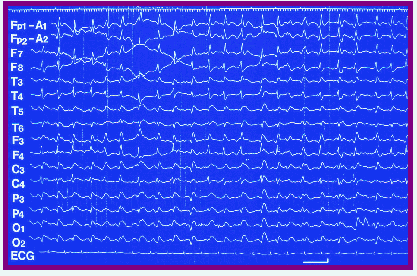
(56歳女性のクロイツフェルト・ヤコプ病患者で見られたPSDです)
画像所見(脳MRI、CT)
脳萎縮が急速に進行し、大脳白質の容積の減少に平行して脳室が著明に拡大してきます。
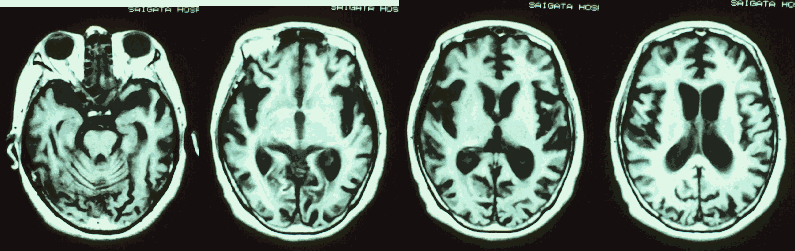
(クロイツフェルト・ヤコプ病の56歳女性の T1強調MRI画像です。)
脊髄液所見
神経特異的エノラーゼ
(neuron-specific enolase: NSE)や、脳由来特殊蛋白(14-3-3)が病初期に上昇します。
*脳由来特殊蛋白
(14-3-3)は、浦堂克美先生(九州大学脳神経病研究所病理学部門)が測定しておらます。
8、クロイツフェルト・ヤコプ病では有効な治療法があるか?
治療法はありません。全身痙攣、ミオクローヌス発作は、対症的に薬剤で抑えられますが、急速に悪化して寝たきりとなり、死亡します。
--------------------------------------------------------------------------------
9、クロイツフェルト・ヤコブ病
クロイツフェルト・ヤコブびょう Creutzfeldt-Jakob Disease ヒトの脳がスポンジのようになってしまう病気。CJDと略す。1920年にドイツの神経病理学者であるクロイツフェルトが、21年に同じくヤコブが報告した。以前は、感染して長期間へたのちに発症するスローウイルスが原因と考えられていたが、最近になって病原タンパク質である
プリオンが関与していることがわかった。この病気にかかると、脳は致命的な損傷をうけ、痴呆状態になっていく。クロイツフェルト・ヤコブ病は世界のどこでもみられる病気だが、患者数は少なく、日本もふくめてどの国でも約
100万人に1人の割合である。50歳以下では、この病気で死亡するケースはほとんどない。70~74歳では100万人に5.9人の割合で死にいたる。死亡年齢の平均は67歳である。50~65歳に多く発症し、性別は関係ない。約90%は1年以内に死にいたるが、なかには1カ月もたたないうちに死亡する例もある。症状
失語、嚥下(えんか)障害、四肢の硬直、顔面筋肉の収縮などの症状があらわれる。その後、合併症によって死亡するケースが多い。およそ
10%が痴呆になり、数年かかって少しずつ脳がおとろえていく。回復した例はなく、治療法もみつかっていない。原因
1982
年、アメリカの神経化学者プルスナーが未知のタンパク質を発見し、プリオンという名をつけた。プリオンは、DNAもRNAももたない特殊なタンパク微粒子である。CJDはこのプリオンの構造が異常になって感染力をもつことから発病すると考えられるようになった。正常なプリオンは健康なヒトや動物ももっていることはわかっているが、プリオンの働きと病気をひきおこす原因はわかっていない。CJDには、孤発性、遺伝性(家族性)、医原性の3種類がある。孤発性
CJD孤発性の
CJDがもっとも多く、人間のプリオン病の中で85%を占める。患者の脳神経(→ 神経系の「神経ネットワーク」)をみると、プリオンタンパクがシナプスに沈着しているのがわかる。このケースでは、プリオンの遺伝子コードが変化する。正常なプリオンは感染性をもたないが、どういうわけか突然変異(→ 遺伝学)をおこし、プリオンの遺伝子が感染性の構造をもつような遺伝コードが指定される。細胞を変質させることは少ないが、まず少量の感染性をもつプリオンタンパク質ができ、これが正常なプリオン分子の配列をかえるようにはたらいて、正常な分子を変質させてしまう。そして、異常なプリオンが一定の量をこえると発病するらしい。この性質は、プリオン以外のタンパク質にはみられない。
遺伝性
CJD遺伝性のものは、プリオンによる病気の
10~15%を占める。変質したプリオンの遺伝子が、世代から世代へと遺伝的につたわっておこる。変質した遺伝子があらわれた家系では、半数がこの病気のために死亡する。医原性
CJD医療によって感染したものである。数は多くないが、角膜移植(→
失明)や硬膜移植(→ 脳)、ヒト成長ホルモンの注射などで感染した例が報告されている。成長ホルモンや角膜、硬膜の提供者がすでに感染していたため発病した。このタイプはごく最近になってわかったもので、研究がすすめられているところである。全世界で2万5000例くらいあると考えられている。1997年3月にWHOは、ヒト硬膜の移植によるCJD感染を報告、ヒト硬膜の移植中止を勧告した。日本でもこれをうけて厚生省は回収命令を通達している。狂牛病と
CJDの関係クロイツフェルト・ヤコブ病は、数が少ないためあまり注目されていなかったが、
1996年、世界をまきこむ騒ぎになった牛のスポンジ状脳疾患(BSE)に似ているため、一躍有名になった。BSEは、1986年イギリスではじめて発見されたウシのプリオン病で「狂牛病」として知られている。狂牛病は、スクレイピーというプリオン病にかかったヒツジの脳や内臓、骨をウシの餌(えさ)にしたために伝染したと考えられている。人間が狂牛病を発病した牛肉を食べたら感染するかどうかについてははっきりわかっていない。1996
年3月末にイギリス保健省は、新しいタイプの致死的なCJD(変態CJD)が10例発見されたと発表した。患者の年齢はすべて42歳以下、脳組織にははっきりした異常がみられ、遺伝的要素はもっていなかった。この患者たちの脳の異常は、それまでのCJDに関する説とは矛盾しているため、新型クロイツフェルト・ヤコブ病とよばれるようになった。家畜と人間の病気の関係は科学的に明らかになっていないが、イギリス政府は、患者が
BSEのウシとなんらかの接触をしたために発病したと発表した。それまでBSEと人間の病気にはいっさい関係はないと否定していた政府が、百八十度態度をかえたというわけである。牛肉からBSEが人間に感染するかもしれないということで、世界じゅうがパニック状態になり、1996年世界各国がイギリスからの牛肉輸入を禁止した。公衆衛生上の見地からすると、CJDやその他のプリオン病について、早急に国際的な医療研究グループの結成がもとめられる。
プリオン病についての英文参考資料 http://www.cdc.gov/ncidod/eid/vol3no2/ricketts.htm