プリオン
Prion目次
Ⅰ.プリオンとは Ⅱ.プリオン病発症のメカニズム Ⅲ.正常プリオンタンパクと異常プリオン蛋白との相違 1)正常プリオン蛋白 2)異常プリオン蛋白 Ⅳ.各具体的疾患について<ヒトのプリオン病について>
<
代表的な動物のプリオン病について>
プリオン病と一括して呼ばれるものは、以前は、伝達性海綿状脳症
transmissible spongiform encephalopathyと呼ばれていた。伝達性というところからもわかるように、感染性がある。かつてはスローウイルス疾患の中に分類されていたが、ウイルスそのものが分離されていたわけではなく、ごく小さな穴のフィルターを通過できるほど小さな病原体ということで、ウイルスと呼ばれていたにすぎない。現在、プリオン病の病原体はプリオンPrionというタンパク質であることが判明している。1970
年代後半プルシナー Pruisnerらは、ネズミの脾臓や脳から、プリオン病の一つであるスクレイピーの感染性を示す物質(scrapie agent)を抽出することに成功した。同様の物質が種々の動物の脳から抽出されたが、これらの物質は核酸を不活化する方法で不活化されず、ウイルスやプラスミドなど従来から知られている病原体と全く異なる性質をもつことが見出された。その後、この物質がタンパク質であることが明らかになり、1982年proteinaceous infectious particleにちなんでPrionと名付けられた。このプリオン蛋白は、宿主(ヒツジ,ウシ,ヒトなど)の遺伝子にコードされている 253 個のアミノ酸より構成されるタンパクで,正常のヒトでも発現している。プリオン病にはいくつかのタイプがある
が、どれもプリオン蛋白が中枢神経に蓄積し、脳が変性して確実に死に至る。この疾患で死亡したヒトや動物の脳にはあちこちに空洞ができている。これがスポンジに似た外観をしているため、「海綿状脳症」という名が付けられた。そのなかで最初に取り上げられたのがヒツジのスクレイピーで、
250年も前のことである。当時、その原因ははっきりとわからなかった。その後、ヤギ、ミンク、シカ、ヘラジカ、ネコ科の動物などによく似た病気が発見されるようになった。狂牛病(牛海綿状脳症)は1986年、イギリスで初めて確認された。ヒトである程度具体的に記述された最初のプリオン病は、パプア・ニューギニアの一部の部族を襲った
クール病である。1957年、アメリカとオーストラリアの科学者が、人肉食の儀式で人間の脳を食べたことが原因であることを明らかにした。それ以後、クール病は事実上消滅した。ヒトで最も拡がっているプリオン病はクロイツフェルト-ヤコブ病(CJD)であり、世界中で感染者は100万人に1人の割合である。他にきわめて珍しいヒトの病気が二つ確認されている。ひとつは Gerstmann-Straussler-Scheinker(GSS)病で、もうひとつは、致死性家族性不眠症である。いずれも一般に遺伝性であり、この家系では、プリオン蛋白をコードする遺伝子が病原性を示すよう変異している。
1.正常なプリオン蛋白が存在する
プリオン遺伝子
…ほ乳類ではありふれた遺伝子である。脳に一番多く発現しているが、他の臓器でも発現している。この遺伝子は正常な個体では少量の正常なプリオン蛋白を作っている。プリオンの遺伝子はヒトでは第20染色体短腕にある。
異常なプリオンが増殖して,海綿状脳症を発症するためには,正常のプリオン遺伝子が存在することが必要である。というのも、正常のプリオン遺伝子をなくしてしまった(ノックアウト)マウスを人工的に作って,そのマウスの脳内に異常なプリオンを接種しても,脳に変化は起こらず,マウスはぴんぴんしているからである。とは言っても,異常なプリオンはプリオン遺伝子をおかしくするわけではない。クロイツフェルト・ヤコブ病の症例中9割では、プリオン遺伝子の異常は見出されない。あとの1割では,遺伝子の異常が見つかるが,このようなケースでも,遺伝子異常は生まれつきにあることがわかっている。従って、
異常プリオンが外からやってきてプリオン遺伝子をおかしくするのではない。
2.正常なプリオン蛋白が何らかのきっかけで異常プリオン蛋白に変わる
1
.遺伝性…プリオン蛋白を作る遺伝子に異常がある場合。(遺伝的)遺伝が関連している時は、アミノ酸の配置異常(置換)によって正常型プリオン蛋から異常型への折り畳みが起こりやすくなっているものと考えられている。
病例:家族性クロイツフェルト・ヤコブ病、致死性家族性不眠症、ゲルストマン・シュトロイスラー・シャインカー病
2
.感染性…以上プリオン蛋白を摂取する、あるいは医療行為等によって異常プリオン蛋白が体内に入ってしまう場合。病例:弧発性クロイツフェルト・ヤコブ病、クールー
3.Bリンパ球が異常プリオン蛋白を運ぶ
スイス・チューリッヒ大学の研究チームは、重い脳神経障害を引き起こすヒツジなどの病気「スクレイピー」が血液感染する原因は、免疫細胞の
Bリンパ球にあるものとする研究結果をまとめた。人間のクロイツフェルト・ヤコブ病でも同様の働きがあると見られ、Bリンパ球の除去が血液感染を防御する手法になる可能性がある。また、スクレイピーの病原体であるプリオンを末梢血に接種しても、
Bリンパ球がないと神経細胞に侵入できないことが動物実験で確認されたため、Bリンパ球がプリオンの運び役になっているとみられる。
4.異常プリオン蛋白が正常プリオン蛋白に働いて、それを異常プリオン蛋白に変えていく
(
三次元で見た正常プリオン蛋白の折り畳み方が変化する・・・連鎖反応)→図A.正常プリオン蛋白の折り畳み変化について
正常プリオン蛋白が異常な形に折り畳まれることで異常プリオン蛋白が生じる
が,この異常な折り畳みはプリオン決定領域(prion-determining region)と呼ばれる部分によって引き起こされる。プリオンは正常蛋白を次々と異常な形に折り畳むため,より多くのプリオンが生じる。プリオンは自然発生するが,通常は害はない。Lindquist
教授は,プリオン決定領域を正常な蛋白に結合させるとプリオンに変化すること,さらには蛋白からプリオン決定領域を除去することで,簡単にこの過程を元に戻すことができることを発見した。共著者であるシカゴ大学院生のNeal Sondheimer氏は「この研究の“売り”はプリオンが蛋白を変化させる要因であるということだ。プリオンは“レゴ(積木の商品名)”のようにお互いに交換可能な部分で構成されている」と述べている。
B.正常プリオン蛋白から異常プリオン蛋白への転換
正常プリオン蛋白がなんらかのきっかけで異常プリオン蛋白に変わったり、外から異常プリオン蛋白(病原体)が接種されると、その異常プリオン蛋白は正常プリオン蛋白に働いて、それを異常型に変えていく。
神経細胞での異常プリオン蛋白伝播経路は、まず、
1分子の異常プリオン蛋白が正常のプリオン蛋白に接触し、これを異常型に変える。次に最初の分子と新しく生まれた異常プリオン蛋白が、2分子の正常プリオン蛋白に接触する。そして、それらが異常型になる。このように、一種のドミノ効果によって異常なプリオンが増殖していくのである。つまり、この転換過程は連鎖反応で、三次元でみたプリオンタンパクの折り畳み方が変化する。タンパク構造の変化と感染疾患との出現との間に、このようなつながりがみられるのは、全く初めてのことである。
この構造の変化は、プリオン病が発症するのは、感染する前に正常なプリオンタンパクが存在する場合に限られるということを意味する。そう考えれば、プリオン病に罹患しているマウスを病原性プリオンでノックアウトマウスを感染させようとしても、常に失敗する理由が理解できる。
5.異常プリオン蛋白が神経細胞内に蓄積する
異常プリオン蛋白は、水に難溶性で、更に分解されにくい性質を持つ
ため、何らかの機序で体内に入っても分解されずに細胞に運ばれる。この外因性異常プリオン蛋白の影響で、細胞由来の正常プリオン蛋白が異常型に変り、異常型と重合する。この結果、異常プリオン蛋白の結晶様構造が細胞内に蓄積し、細胞死(アポト-シス)が起ると考えられている。(プリオンは生体に存在するタンパク質に由来するため、感染しても免疫反応が起らない。)6.発症
*
ヒトでは第20番染色体に存在するプリオン遺伝子が産生する分子量3.3-3.5万の糖蛋白であって、細胞膜に結合して存在する*
プリオン遺伝子と正常なプリオン蛋白は健常な動物にも存在する*
プリオン遺伝子は哺乳動物から酵母にいたるまで見いだされている普通の遺伝子である(←
Prusinerのほか、Charles Wiessmannが率いるグループが実験を繰り返し、数種類 の動物でプリオンタンパク遺伝子の配列を明らかにしようとした。その結果、ウシのプリオン蛋白とヒツジのプリオン蛋白とを比較することが可能となり、(蛋白を構成している)計264個のアミノ酸のうち、わずか7個が異なるにすぎないことがわかった。また、人間には安心材料になると思われるが、ヒトではウシのプリオン蛋白と異なるアミノ酸が30個は下らないことがわかっている。それでも、Prusinerは次のように警告している。「重要な領域でウシプリオンタンパクのある部分がヒトプリオン蛋白と同じ配列にそっくり変わってしまうかもしれません。そうなれば、全アミノ酸配列の単純比較から考えられるよりも、危険は大きくなります」)*
蛋白分解酵素で分解される*
タンパクの高次構造は、αへリックス構造
〇正常プリオン蛋白と異常プリオン蛋白では、アミノ酸の一次配列は同じ。
(
正常型)プリオン蛋白は脳や神経の細胞膜表面に存在する、分子量3.3-3.5万の糖タンパク質である。粗面小胞体で産生されたプリオン前駆タンパク質はゴルジ装置で糖を付加され、プリオン蛋白となって細胞膜表面に運ばれる。正常の細胞では、エンドサイトーシスによってプリオン蛋白が細胞内に再び取り込まれ、加水分解される。プリオン病では、細胞内に取り込まれたプリオン蛋白が完全に加水分解されず、分子量2.7-3.0万の異常プリオン蛋白に変わる。正常プリオン蛋白の一部分が加水分解されて異常型に変るため、正常型と異常型の一次構造は全く同じである。しかし、正常型と異常型の二次構造は異なり、この二次構造の差が異常型プリオン蛋白の病原性を決定づける。)
〇ヒトのプリオンの正確な構造は
Science233:364(1986)に最初の報告があり、分子量27000 で,253のアミノ酸残基からなる。プリオンの遺伝子は、ヒトでは第20染色体短腕にある。現在、
Human Frontierプロジェクトの一部に特殊班を設け、プリオンタンパクのどの部分が重要であるかという問題に取り組んでいる状態で、今、正常マウスのプリオン遺伝子からさまざまな部分を切り取っている段階である。これに、プリオン病に罹患したマウスの病原性プリオンを接触させ、どの部分を切り取れば感染しやすくなるか、あるいは感染しにくくなるかをみようとしている。これまでに、プリオンタンパクの一端にある約60個のアミノ酸が、感染しやすさには関係がないことが明らかになった。
プリオン病の主なものとしては、羊のスクレイピー、人のクロイツフェルト・ヤコブ病(CJD)とクールー、牛の狂牛病(正式には牛海綿状脳症:BSE)などがある。→分類表
<ヒトのプリオン病について>
CJD):遺伝性+外的要因によるA.弧発型
CJDなぜ
PrPscが発生したのか全くわからないもの。実際には、このタイプが全体の90%以上を占めている。特発性クロイツフェルト・ヤコブ病(CJD)、医原性クロイツフェルト・ヤコブ病、異型(新型)クロイツフェルト・ヤコブ病(nv-CJD)がある。・特発性クロイツフェルト・ヤコブ病
*
症状と診断*40
~50歳の初老期に発症することが多いのですが、80歳以上で発症することもある。怒りっぽくなったり逆に無関心になるなどの性格変化や疲れやすさなど のはっきりしない初期症状で始まることが大半ですが、視力障害や歩行障害が先行することもある。数週間のうちにおよそ考えられる限りの多彩な神経症状・精神症状が入れかわりたちかわり出現する。やがて、この病気に特徴的なミオクローヌスという筋肉のぴくつきが現れる。ミオクローヌスの出現と同時期には、脳波検査で非常に特徴的な周期性同期性放電(PSD)という波がみられるようになる(脳波所見)。脳脊髄液検査で14-3-3蛋白という特殊なタンパク質が検出されることもある。この頃には、ほとんど外部からの刺激には反応できなくなり、数ヶ月の間に寝たきりになってしまう。寝たきりになってから頭の写真をとると、他の病気では考えられないほどの速さで脳の萎縮(痩せ)が進んでいくのがわかる。(写真→正常脳,CJDの脳)これらの検査からクロイツフェルト・ヤコブ病の診断を行う。
*
治療*治療法はない。ミオクローヌスやけいれん等に対してクロナゼパム、バルプロ酸ナトリウムなどの抗てんかん薬が使われる場合もあるが、病気の進行が早いため、使用する機会は実際にはほとんどない。多くは1年前後で肺炎などの合併症で死亡する。寝たきりになってからの生存期間は栄養管理や合併症の治療がどの程度うまくいくかにかかっている。死後の脳の多くは正常の半分以下にまで縮み、脳のあらゆる場所で神経細胞が抜け落ち、スポンジ状に変化している。(
写真)・医原性クロイツフェルト・ヤコブ病
ヒト乾燥硬膜移植後
CJDは1979年から1987年までに脳外科手術の際に硬膜移植を受けた患者さんに発生した。脳外科手術を行う際には、頭蓋骨を取り、その下にある硬膜を切開して脳に到達するが、手術後、切開した硬膜を元通りに縫いあわせるための材料が必要になる場合がある。この時、ヒトの遺体から採取した硬膜が使用されていた。現在は『ゴアテックス』という樹脂製の人工硬膜が使われている。少なくとも1980年代後半までは、日本ではドイツのBブラウン社が開発した『ライオデュラ(Lyodura)』という輸入製品がほぼ独占的に使われていたのだが、この製品を移植されたと考えられる患者さんにきわめて高い率でCJDの発症がみられた。 (1985年に他社から発売された『ツトプラスト(Tutoplast)』というヒト乾燥硬膜では全くCJD発症例が報告されていない)。2000年3月までの時点で硬膜移植歴のあるCJD患者数は実に67名に上っている。概算では、弧発型CJDの数千倍の発病率になる。Bブラウン社の製造過程(ドナーの選別がきわめていい加減に行われていた、ドナー記録を全く残していなかったなど)や販売方法(未処理死体硬膜の使用禁止命令後の密売など)に数々の問題があったことが判明し、1996年には同社は販売中止の処分を受けている。このほかに角膜移植や脳深部電極による脳波記録、下垂体由来の生長ホルモン製剤によっても感染したという報告があるが、下垂体生長ホルモン製剤は1985年以後遺伝子組み替えによって製造されているので安全である。・異型クロイツフェルト・ヤコブ病
1994
年になって、イギリスで異常に若い年齢(通常は初老期発症が多い。後述)で発症したCJD患者が見つかり、新変異型CJD(new variant CJD:nv-CJD)と名づけられた。発症年齢だけでなく、検査所見・病理所見いずれも古典的なCJDとは異なったもの(前出のクールーによく似ている)であり、状況証拠がそろううち、このnv-CJDがBSEのヒトへの伝染の結果である可能性が極めて濃厚になってきた。 (ロンドン大学のジョン・コリンジらの報告が有名である)。*
発症年齢層*1996
年に変異型CJDが報告された時、もっとも若い患者は16才だった。1997年5月から3年間にわたって16才以下の子供について変異型CJDの積極的な調査が行われた。対象となったのは進行性の知的および神経障害を示した子供である。この症状が疑われた885名の患者の中から、2名が変異型CJDと診断され、1名がほぼ確実例とみなされた。確定診断例は14才の女子と15才の男子で、いずれも死亡している。ほぼ確実例は12才の女子である。この成績に対して3年間では期間が短すぎるとの意見も出ている。一方、これまで変異型CJDは若い人が中心だったが、昨年は74才の人が死亡した。このことは、当初予想されたのよりもはば広い年齢層で患者が発生する可能性を示している。*
発生状況*変異型
CJD患者の発生状況 Lancet 356, 482(8月5日号)によれば、英国での患者数は2000年6月現在で75名である。内訳は1994(8)、1995(10)、1996(11)、1997(14)、1998(16)、1999(16)、2000(0)だ。そのうち69名が死亡している。59名は病理学的に変異型CJDと確認されており、ほかの16名(このうち6名は生存しています)は、ほぼ確実例とされている。この内訳を見ると毎年、患者数は23%増加しており、死亡数は33%増加していることになる。絶対数は低いものの、増加している点に関心が寄せられている。*
今後の見通し*ところで、
BSE発生の1980年から変異型CJDが初めて見つかった1996年までに、英国では75万頭の狂牛病感染ウシが屠殺されたと推定されている。そして、最大50万人の変異型CJD患者が発生するとの予測がなされている。この予測が下方修正された。今年になってオックスフォード大学のニール・フェルグソン
Niel Fergusonのグループが試算した結果、最大13万6000人という推定が出された。かつての予測値よりはかなり少なくなったわけである。 実際には潜伏期など、いろいろな要因が不明なために、最終的な予測はできない。平均寿命よりも潜伏期が長くなければ、6000人以上になることはないだろうという推測もされている。
B.遺伝型CJD
20
番染色体上にあるPRNPに変異(遺伝子異常)を持っているためにPrPscが作られてしまう疾患群。重要なのは、PRNPに変異があるからといってそれが即プリオン病の発病や遺伝性につながるのではなく、むしろプリオン病を発病するような変異を持つことの方がはるかに稀な出来事であるという点である。PRNPに変異があっても、圧倒的大多数の人では何の障害も遺伝的問題も発生しない。家族性クロイツフェルト・ヤコブ病がある。・家族性クロイツフェルト・ヤコブ病
20
番染色体上の200番遺伝子(codon200)から作られるアミノ酸に異常がある(正常はグルタミン酸であるがそれがリシンに変わってしまう)ものが代表である。弧発性CJDと区別のつかない病状・病理所見を示す。リビアはCJDの多発地帯で、かつ羊の眼球を食べる習慣があったため、当初はスクレイピーの感染が疑われたが、その後の研究で遺伝子異常が原因であることが判明した。その他に180番遺伝子、210番遺伝子、232番遺伝子などの異常による遺伝性CJDが知られている。現在日本には8名(登録者のみ)の患者さんがいる。
*
発症地区*パプアニューギニアの高地に、昔から死んだ人の葬送の意味から、肉親による死体の食肉の風習があり、その地方に風土病的にクールーという病気が知られていた。
*
症状*発病すると
急速に痴呆、精神異常、ミオクローヌスという不随意運動、さらに植物状態から死に至るという病気で、脳は海綿状に破壊されるといわれる。スクレーピー、狂牛病、クールー、ミンク脳症、クロイツフェルド・ヤコブ病、いずれも病像と病理所見は酷似していると言われる。*
特徴*第
2次大戦後、石器時代さながらの生活を営んでいたフォア族に奇妙な病気が発見され た。現地語で震えを意味するクールーという病気で、患者の多くは若い女性で震えなどの症状から麻痺が起こり半年前後で皆死亡した。クールーの最初の患者は
1920年代に出現したと推測されている。おそらくCJDの患者で、それが食人の儀式で広がった。食人は同じ部族内の風習で、通常は親族、特に両親が死亡した際に敬意と感謝の意を示すために行われた。クールーは死者の脳のようにプリオンが多く含まれる組織を食べたことのほかに、調理の最中に傷口などからプリオンが感染したことが原因と考えられている。そのため女性に多く起こり、発生のピークとなった1960年代には、女性の最大の死亡原因となり全部で2千人以上が死亡した。食人の風習は
1950年代終わりにかなり急速になくなりクールーの発生も終息していった。しかし、現在でも食人の習慣が存在した時期より前に生まれた人の間で患者が毎年数人出ている。潜伏期は40年を越すことになる。*伝染性と予防*
プリオンタンパクは、他のすべての生物が当然持っている遺伝子情報である核酸を持たな い。しかも、極めて安定した蛋白質で少々加熱したり、放射線や紫外線を当てたり、通常の消毒薬に浸しても、その伝染性は失わない。つまり感染した動物の生肉を食べるのみならず、感染した動物の肉を煮たり、焼いたりして食べても、プリオン病にうつる可能性があるということである。このようにプリオンの完全な不活化はおそらく不可能であるため、
予防法としてはプリオン病の可能性のある食品は食べない、触れないことである。
*
今後の課題*公衆衛生の立場ではプリオンを短時間に高感度で検出する方法の開発が緊急の課題となっている。これにより潜伏期の患者も含めて急速に診断することが可能となり、また血液、医薬品、食品などのプリオン汚染の検出対策が可能となる。効果的な不活化方法の開発は医療器具などを介する医原性
CJDの予防のためのきわめて現実的な問題である。しかし、プリオンの完全な不活化はおそらく不可能であるため、放射能などの場合と同様に安全限界を知ることが必要である。それには発病機構の解明と発病に必要なプリオン量を知ることが必要である。発病したら確実に死亡するプリオン病の治療法の開発もまた緊急の課題である。Fatal familial insomnia):遺伝性による
世界中にほとんど例がなく、頑固な不眠と多彩な自律神経症状を示し、やがて昏睡、死に至る苦痛に満ちた遺伝病である。
→用語集
<
歴史>1986年 Lugaresi等による研究
Little等による家系の報告
1995年 立石等によるマウスにおいてFFIを遺伝させることに成功
現在まで約30件の報告がある。このようにして、FFIは大脳のアミロイドーシスのグループに分類されるようになった。
<発病の原因>FFIは常染色体優性遺伝である。
プリオン蛋白遺伝子の
の二つの変異が条件である。
これらには
ばらつきがある。診断には、
「不眠、自律神経系の過度の活動、家族で発現、昏睡後死亡」
が最小限基準
。
<
症状>・進行するに連れて、構語障害、四肢腱反射亢進、ミオクローヌスを呈するようになり、まったく反応の得られない無動昏迷状態を経て、発症後1~3年で死に至る。
3
.ポリソムノグラフィ…正常な睡眠周期は失われている。睡眠覚醒機構の障害。4.病理学的見地…視床前核と背内側核にグリオーシスを伴う両側性の著しい神経脱落→その後、皮質―皮質下の海面状変化
赤い部分が代謝活性な部分。
進行期である
Case1とCase3では、視床だけでなく大脳の広範な代謝低下が見られる。病初期であるCase4では、視床前部の代謝低下のみで、皮質―皮質下の代謝は保たれている。→
視床
<不眠の原因>
紡錘波の消失と、覚醒への抑制機構の障害のため
。
<自律神経機能障害の原因>
症状より、
FFI
では睡眠覚醒機能の障害がある一方、視覚、聴覚、体性感覚による誘発電位は正常に保たれている。また、自律神経機能については、
副交感神経機能は正常である一方、交感神経活動が異常亢進している。
しかし、この交感神経機能障害の原因が
のどちらによるのかはわかっていない。
ここに、FFI患者の生態リズムの側面について検討したものがある。
血中の成長ホルモン、プロラクチン、
ACTH、コルチゾール、カテコラミン、などの分 泌リズムも正常パターンから逸脱している。特に、アドレナリン、ノルアドレナリンは異常高値が終日持続している。
*
定義*ゲルストマン・ストロイスラー・シャインカー病(GSS)は家族性プリオン病で、進行性小脳失調症、あるいは痙性対麻痺と痴呆を主症状とし、数年後に無動・無言状態となる。神経病理学的には大脳と小脳にアミロイド斑を多数認め、プリオン蛋白から構成されている。プリオン蛋白遺伝子の変異を必ず伴っている。
プリオン病のなかでも、遺伝性疾患であるが、遺伝的に作られた異常型プリオンにも感染性はある。
*
病因*プリオンタンパク遺伝子の
特定部位の変異がGSS発症と深くかかわっている。現在のところGSSの全例で プリオン蛋白遺伝子の変異が明らかになっている。変異の浸透率も高く、90%以上と報告されている。病理像の特徴は、 全症例にプリオン蛋白からなるアミロイド斑が認められる(GSSはアミロイドーシスに含まれる)ことで、海綿状態やグリオーシスは症例により違いがある。*
症状*プリオン蛋白遺伝子の各変異ごとに臨床症状が異なっている。
日本で見られるGSSで、最も多いのは
進行性の小脳症状を主徴とする病型で、コドン102の変異を示す。小脳症状として失調性歩行、四肢の失調、構音障害、眼振などで 初発し、徐々に進行する痴呆が現われ、5~10年後には寝たきりとなる。次に多いのは痙性麻痺型である。下肢の深部腱反射亢進、
病的反射、痙性歩行などで始まり、徐々に痴呆症状が加わってくる。コドン105の変異が特異的である。その他、痴呆で始まり、 徐々に進行する病型もある。ミオクローヌス*は稀か、ほとんどみられないことがCJDと異なる。病初期には鑑別診断が困難なことがあり、注意を要する。*
治療 *疾患を回復し、進展を阻止又は遅延させる有効な治療法は知られていない。痙性症状が強い症例には抗痙縮剤(バクロフェン、
ダントロレン、ジアゼパム)などで強い痙直を取り除くことにより看護が容易になる。栄養の補給、拘縮や褥瘡の予防、呼吸器や尿路感染の防止が必要となる。
*
予後 *予後は不良である。GSSの全経過は平均して約6年である。
発病後2-10年に全身衰弱、呼吸麻痺、肺炎などで死亡する。
*
現状*年間のクロイツフェルト・ヤコブ病を中心としたプリオン病は、100万人に1人の割で発症するが、そのうちゲルストマン・ストロイスラー・シャインカー病は、プリオン病の
10~15%を占める。男女間に差はない。発症年齢は 40~50歳代が多く、若年発症(20~30歳代)もみられる。
<代表的な動物のプリオン病について>
Scrapieスクレイピーは
1732年に英国で最初に報告されたヒツジの遅延性脳症である。1930年代に、発病した羊の脳を健康な羊に接種すると感染が成立し、発病することがわかり、伝染性の病気であることが確かめらた。主な症状は中枢神経系が特異的に侵されることによる神経症状
である。発症したヒツジは動きが鈍くなる、激しい掻痒感のために体をこすりつけて毛が抜ける、震えを起こす、足を高く挙げて歩く、立てなくなるなどの運動障害を引き起こし、最終的に死に至る。Scrapieという名前は、ヒツジが体をこすりつける(scrape)することに由来する。発症したヒツジの脳を解剖すると、神経細胞の死滅と共に、脳幹部の空胞化、アミロイド用繊維の形成が認められる。ヒツジは牧場内で出産し胎盤を放出する。ごく希に自発的に生じたスクレイピーを持つヒツジの胎盤により汚染された牧草を、他のヒツジやオオシカ、ヤギなどが食べることにより、プリオンが水平伝播して病気が広がった可能性がある。
世界中でスクレイピーの存在しない国はオーストラリアとニュージーランドだけだといわれている。日本もフリーだったが
1974年にカナダから北海道に輸入した羊により持ち込まれ、東北、関東、九州にも拡がった。ただし日本では羊の飼育頭数が少ないので、大きな流行にはなっていない。
正式には、
牛海綿状脳症(Bovine Spongiform Encephalopathy:BSE)という。1986年にイギリスで始めて存在が確認されたが、前年からスクレイピー様の症状をもつ牛が散発的に観察されていた。狂牛病の牛は恐怖に怯えたり、攻撃的になったりする異常行動で発病し、やがて体が震えて立てなくなる。物音や触覚刺激に過敏に反応するようになり、病状が進行して数ヶ月以内に屠殺されることになる。脳組織は海綿状(スポンジ状)を呈し、星状細胞が増殖し、神経細胞の脱落が見られ、スクレイピーやCJDの病理所見とほぼ同じ像を呈する。潜伏期は2年以上ある。狂牛病は、羊の海綿状脳症であるスクレイピーに感染した羊の内臓、くず肉がたまたま子牛の成長を促進するための濃厚飼料として使われたことにより牛の間で広まったと考えられる。
1986年以後狂牛病の牛は増え続け、1992~93年にピークに達し、年間3万7千の牛がこの疾患に罹患した。狂牛病の牛の誕生年を調べると、1987年に誕生した牛が最も多いことが判明した。羊のスクラピーが牛に感染した可能性が指摘されたため、1988年7月英政府は、牛や羊の内臓を牛の飼料(meat and bone meal=MBM)として用いることを禁止した。しかし、既に出回っていたMBMの一部はそのまま使われ、MBM禁止後に生まれた牛に狂牛病が出た。また、餌からの感染は牛にとどまらず、ロンドンの動物園ではウシ科の数種の野生動物が餌から狂牛病に感染した。英国の家猫もキャットフードからこれまでに
77頭が感染した狂牛病の原因になった餌は日本や米国でも用いられていたが、幸い狂牛病は発生しなかった。
<参考文献等>
http://www.google.com/ http://square.umin.ac.jp/~massie-tmd/bsecjdexp.html http://square.umin.ac.jp/~massie-tmd/bsecjdexp.html http://naoru.com/kuroituferuto.htm http://biokagaku.com/NR/2000/Jan00/J013100.htm http://jhfsp.jsf.or.jp/pub/pub97/prion.html http://www.kyushu-id.ac.jp/~hoken/siori/99Purion.htm http://jhfsp.jsf.or.jp/pub/pub97/prion.html致死性家族性不眠症
用語
http://ss.niah.affrc.go.jp/bse/term-s.html英語
http://www.ncbi.nlm.nih.gov/htbin-post/Omim/dispmim?600072→
英訳「第
10巻 睡眠とその障害」監修 高倉公朋・宮本忠雄、発行者 中尾俊治、発行所 株式会社メジカルビュー「睡眠学ハンドブック」
1994 発行者 朝倉邦造
<製作者>
99061
砥上 妃美子 ,99077 平田 留美子 ,99081 松野 文子 ,99082 松本 美根子99083
道又 潮香 ,99089 山田 友子 ,98034 塩沢 理世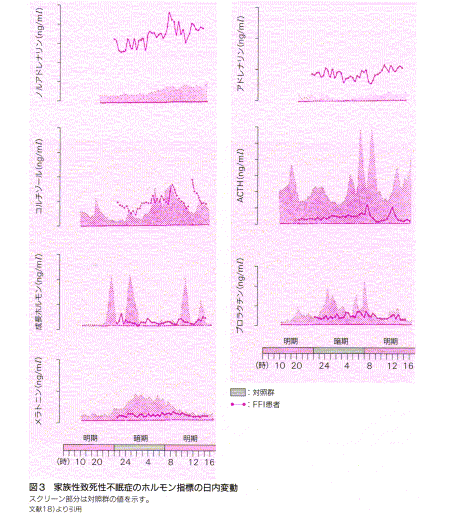{kind=link}